Function of the program: The program reads in spherical virus electron-density maps and project the densities along defined radial vectors onto a sterographic sphere. The projected density can be represented as contour lines or as gradient of colors. The program can also read atomic coordinates from a PDB and project chosen residues as roadmap onto the same sterographic sphere. The residues can be labeled and colored in various way. The program is still actively developped. If you have any problem of using it. Please contact the author at Chuan (River) Xiao (cxiao@utep.edu)
Citation of the program: Please cite the paper in your publication using "Xiao, C. and Rossmann, M. G. (2007). "Interpretation of electron density with stereographic roadmap projections." J Struct Biol 158(2): 182-7. PubMed PMID: 17116403;PMCID: PMC1978246; Download PDF.
Download of the program
Latest version: major changes with support of the APBS potential maps, tested but might have bugs, please report if you encount bugs.
Stable versions: match the publication with minor changes.
V4.5 64bit; V4.5 common linux;
Earlier versions:
V4.2 64bit; V4.2 common linux; V4.0 64bit; V4.0 common linux;
V3.2 common linux; V3.1 common linux; V3.0 common linux; V2.9 common linux;
Testing files with examples:
Purpose of the program: Studies of receptor binding to the surface of parvoviruses presented a problem of how to visualize the interaction between the two complex molecular surfaces. The program RIVEM (Radial Interpretation of Viral Electron density Maps) was developed to project an asymmetric receptor density radially onto a unit sphere using spherical coordinates. The stereographic diagram of the projected densities allows a clear visualization of the receptor footprint on the icosahedral virus surface. A spherical "roadmap" function was incorporated into RIVEM so that virus surface residues were projected within the same unit sphere and therefore superimpose onto the projected receptor densities. The program offers a powerful way to visualize and study the correlation between atomic structures and electron density maps in a generic variety of model systems.
* Input map can be ommitted so that it will only generate the sperical roadmap. But -O option must be set.
| Options | explanations |
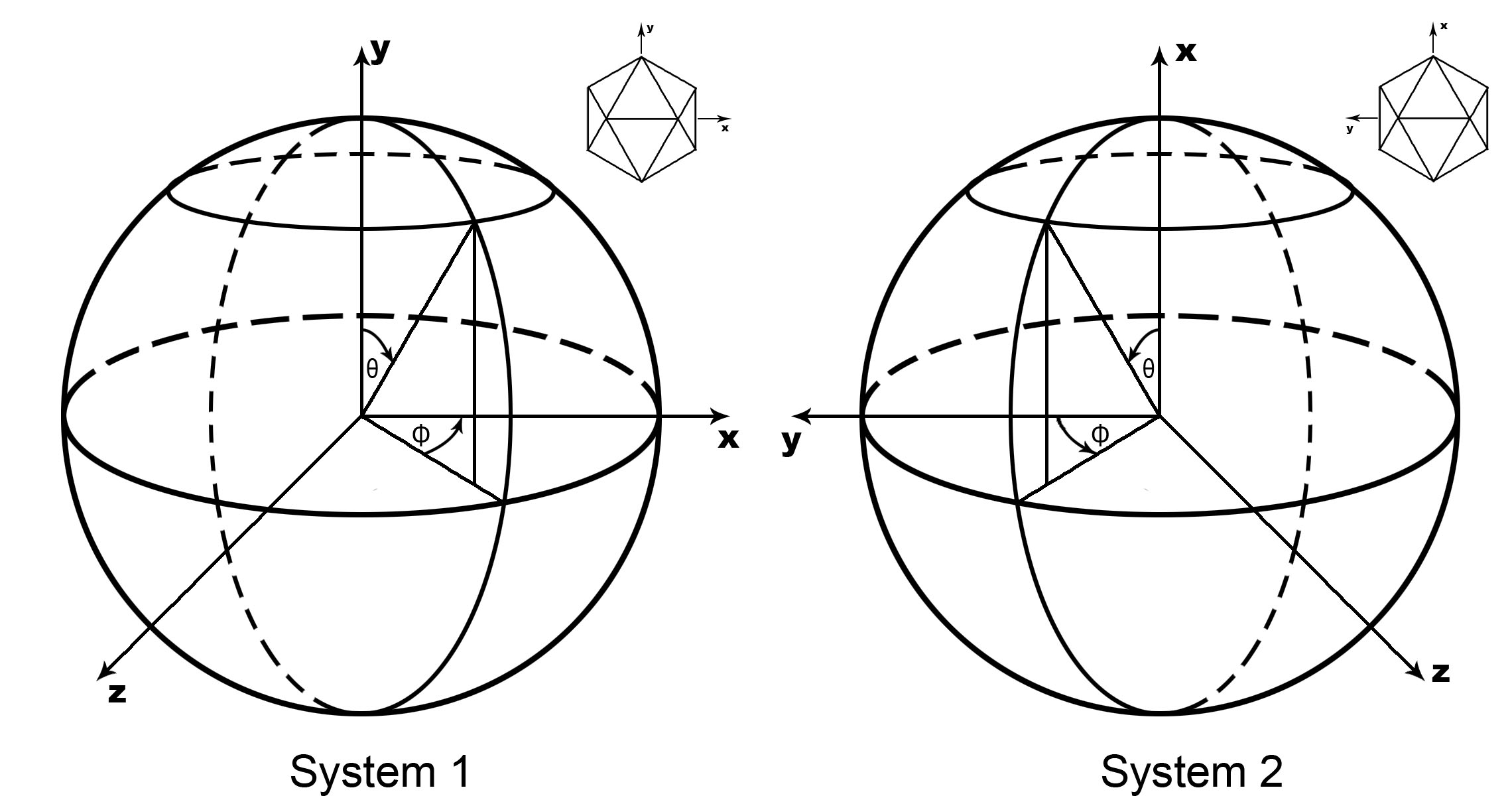
| -S polar | Define the polar system,default is 1 polar = 1: Theta(Psi) is rotate from X Phi is rotate from Y toward Z polar = 2: Theta(Psi) is rotate from Y Phi is rotate from X away from Z 2 is the same as Rossmann&Blow,1962 |
| -Pbeg[in] begPhi | Plot start with begPhi,default 0.0 |
| -Pend endPhi | Plot start with endPhi,default 180.0 |
| -Tbeg[in] begTheta | Plot start with begTheta,default 0.0 |
| -Tend endTheta | Plot start with endTheta,default 180.0 |
| -D delta_angle | Define the increment step of angle,default 1.0 degree |
| -Xbeg[in] begX | Plot start with begX in Angstrom, default -3.402823E+38 |
| -Xend endX | Plot start with endX in Angstrom, default 3.402823E+38 |
| -Ybeg[in] begY | Plot start with begX in Angstrom, default -3.402823E+38 |
| -Zbeg[in] begZ | Plot start with begX in Angstrom, default -3.402823E+38 |
| -Zend endZ | Plot start with endX in Angstrom, default 3.402823E+38 |
| -O | Turn off map countour plotting, only plot roadmap, map filename should be omitted, otherwise it will be regarded as the PDB input |
| Options | explanations |
| -E add_map_n | Define the number of additional maps, mimimum is 1,maximum is 9 default is no additional map will be read |
| -eX mapfilename | Read-in the additional maps to superimpose on the original map, X support from 0 to add_map_n-1 if you have 1 additional map, X should be 0 (C language style) |
| -A[X] average | Use the NCS to average the map (average=1) or apply values from one asymmetric unit to other asymmetric unit(average=2) to the other asymmetric unit. X: if exist corespondent to the additional maps |
| -I[X] | Use icosahedral recalculated radius instead of the original radius X if exist coorespondent to the additional maps, this option require -m to input the icosahedral matrices |
| -R[X]fix fixR | Define the perticular radius that need to plot,see -Rmode,default 0.0A X if exist coorespondent to the additional maps |
| -R[X]beg[in] begR | Define the starting radius in A for density projection,default 0.0A X if exist coorespondent to the additional maps |
| -R[X]end endR | Define the end of the radius in A for density projection, default is the maximum diagonal radius of the map X if exist coorespondent to the additional maps |
| -R[X]step delta_R | Define the increment step of the radial projection, default is the map voxel size, X if exist coorespondent to the additional maps |
| -R[X]mod[e] Rmod | Define the radial interpretation mode,default is 1
* be careful to use and interprete the plot that use Rmod=0 because the sum of the density will have much larger dynamic range than the original map. Choose the contour settings of -C carefully. Using small delta_R will further increase the range. It will also affect the -c color_method option 4 and 5 that color the residues with density map |
| -C[X] contour_start contour_end contour_step line_type color | Contour the map X in certain line type and color,
X corespond to the additional map number Line_type
|
| -p PDB_filename | Give the name for the PDB file for the Roadmap |
| -a maximum_atom_number | Define the maximum_atom_number,default is 500000 |
| -B border_width border_color | Define the roadmap outline line width and border color, default line width will be automatic determined by the program based on the angular step, delta_angle(see -D); color option see -C, default color will be black(16) |
| -b add_atom_move | add additional atom movement in A, default 0.0, default atom radius is van der Waals radius plus the affect of temperature (B) factor |
| -l label_size label_color | Turn on label of the residues on the roadmap and define the label size and label color; label color option see -C; when label_size equal 0 the program will automatic determine it based on the angular step, delta_angle(see -D) |
| -rmin[imum] r_min | Define minimum radius for atom coordinates, atoms with radius smaller than r_min will not be read in |
| -rmax[imum] r_max | Define maximum radius for atom coordinates, atoms with radius larger than r_max will not be read in |
| -rfix r_fix | Define the perticular radius to find the closest atom |
| -rmod[e] r_mod | Define the mode of the roadmap, default is 1
|
| -xmin[imum] x_min | Define X minimum for atom coordinates, atoms with X smaller than x_min will not be read in |
| -xmax[imum] x_max | Define X maximum for atom coordinates, atoms with X larger than x_max will not be read in |
| -ymin[imum] y_min | Define Y minimum for atom coordinates, atoms with Y smaller than y_min will not be read in |
| -ymax[imum] y_max | Define Y maximum for atom coordinates, atoms with Y larger than y_max will not be read in |
| -zmin[imum] z_min | Define Z minimum for atom coordinates, atoms with Z smaller than z_min will not be read in |
| -zmax[imum] z_max | Define Z maximum for atom coordinates, atoms with Z larger than z_max will not be read in |
| -c color_method | Turn on coloring the residues on the roadmap
* Only the original input map density can be used to color the
roadmap residues(color_method 4 and 5), all the additional map can only be plotted
with contoure lines using -C
** The name of the PDB file that controls the residue color needs to
be started with letter instead of number. Otherwise it might be read as
a numbered option 1 to 5 |
| -G color_mid_point | Define the color middle point for RGB color gradient option -G and -g are related to how to make a RGB gradient The formula of a RGB gradient is the same as described in VMD http://www.ks.uiuc.edu/Research/vmd/vmd-1.7.1/ug/node76.html |
| -g color_min | Define the color minimum for RGB color gradient option -G and -g are related to how to make a RGB gradient The formula of a RGB gradient is the same as described in VMD http://www.ks.uiuc.edu/Research/vmd/vmd-1.7.1/ug/node76.html |
| -dmin d_min | Set the lowest plot value for the RGB gradient, related to the color_method, it can be the smdallest radius or the lowest density default will be lowest radius calculated during the plot or the negative maximum absolute value of the lowest and highest density (-MAX(abs(density_min),abs(density_max))).For density,the input d_min smaller or equal to zero |
| -dmax d_max | Set the highest plot value for the RGB gradient, related to the color_method, it can be the largest redius or the highest density default will be the highest radius calculated during the plot or the positive maximum absoluted value of the lowest and highest density (MAX(abs(density_min),abs(density_max))). For density, the input d_max must larger than zero |
| -s plot_axis | Plot the icosahedral axis with plot_axis_color.
plot_axis default is 0, not ploting
the axes are defined in the matrix_file input by option -m
|
| -sc axis_color | define axis_color, default is 16(black) see -C for color code |
| -ss symbol_size | define symbol_size, default is 6, can be 1 to 12 |
| -sb asu_border_color | define asymetric unit border color, default is 16 (black), see -C for color code |
| -sl asu_border_linetype | define asymetric unit border_linetype, default is 3 (solid,width 1.0), see -C for linetype definition |
| -m matrix_file | Read in the matrices for the PDB and the maps (include additonal maps),
the matrix_file should be in CNS ncs.def matrix format, see
examples* *Both NCS and map (include additional maps) rotational -translational matrices are read from the matrix_file |
| -M matrix_max | Define the maximum matrix number, default is 100 |
| -N axis_number axis(1)_theta axis(1)_phi axis(1)_fold axis(1)_size
axis(1)_color ...... axis(axis_number)_theta axis(axis_number)_phi axis(axis_number)_fold axis(axis_number)_size axis(axis_number)_color | Draw additional defined symmetry symbols axis_number define how many axes. Each axis is controled by five parameters: theta, phi, fold, size and color; theta, phi should be in the plot range, fold options are 2,3,4,5,6. Check -C for color codes |
| -L line_number line(1)_theta_start line(1)_phi_start line(1)_theta_end
line(1)_phi_end line(1)_color line(1)_type ...... line(line_number)_theta_start line(line_number)_phi_start line(line_number)_theta_end line(line_number)_phi_end line(line_number)_color line(line_number)_type | Draw defined additional line (great cycle curved line) by inputting start and end polar angles and line type. The polar angle should be inside the plotting range. Check -C for color codes and line types |
| -F label_number label(1)_theta label(1)_phi lable(1)_size label(1)_color
label(1)_text ...... label(label_number)_theta label(label_number)_phi label(label_number)_size label(label_number)_color label(label_number)_text | Put a text label at input polar angle, the angle need to be in the plot range. No space is allowed in label_text, use quotation marks. Check -C for color codes |
| -v verbose_level | Define the verbose level. Default verbose_level=1
|
| -h | Print the help manual |
rivem -S 1 -l -v 1 -c 3 -dmin 110.0 -dmax 150.0 -s 2 -m ncs.def -D 0.1 -Pbeg 62.0 -Pend 118.0 -T beg 55.0 -Tend 95.0 -p CPV.pdb -O color_by_radius.ps
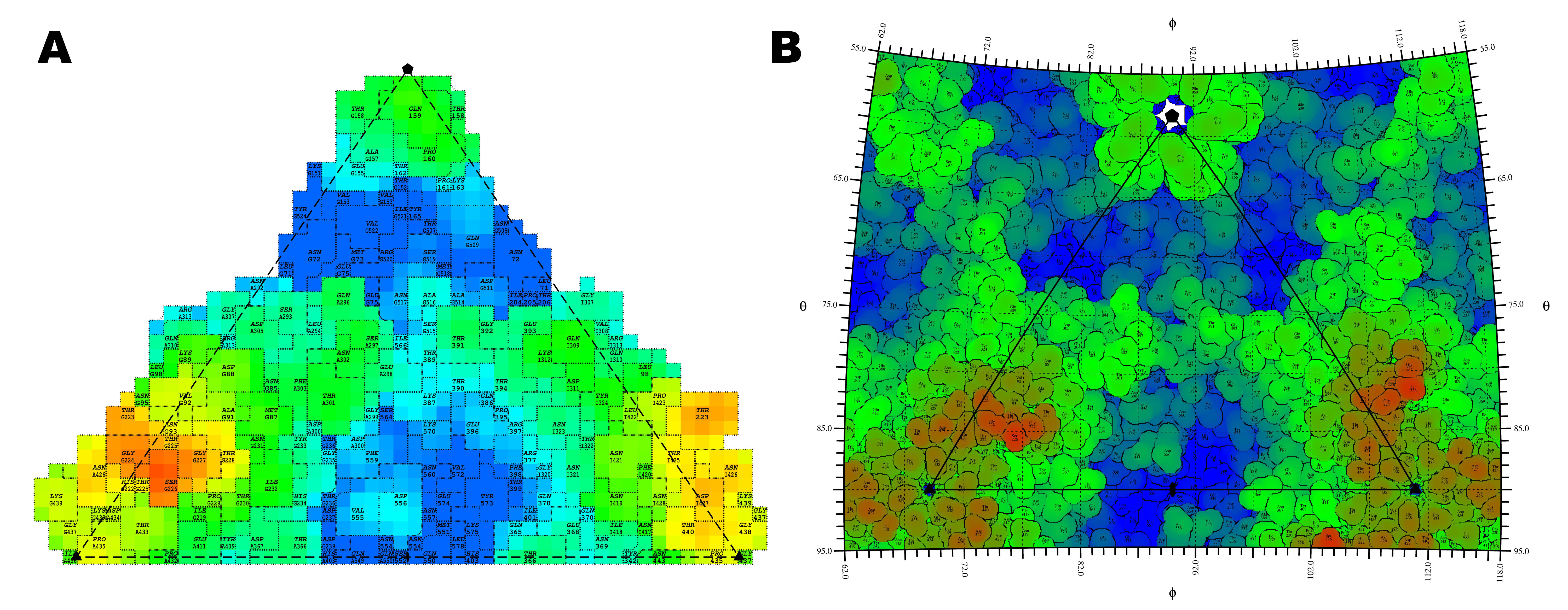
Roadmaps of CPV.
A. CPV surface residue plot generated
by the ROADMAP program (Chipman, M 1993). Surface residues are colored according to
the Z value of the atom positions in the Cartesian coordinate system. B. CPV surface
residues plotted by the RIVEM program. Surface residues are colored based on the maximum
radial distance Rmax of the atom to the virus center in a spherical coordinate system.
rivem -S 1 -C 25.0 70.0 5.0 3 1 -c color.pdb -l -v 1 -s 2 -m ncs.def -D 0.1 -Rbeg 140.0 -Rend 200.0 -Pbeg 62.0 -Pend 118.0 -Tbeg 55.0 -Tend 95.0 -p CPV.pdb diff.xplor asym_colorresidue.ps
rivem -S 1 -s 2 -v 1 -m ncs.def -D 0.1 -C 25.0 70.0 5.0 3 1 -C -70.0 -25.0 5.0 5 0 -c 4 -E 1 -e0 diff.xplor -R0beg 140.0 -R0end 200.0 -C0 25.0 70.0 5.0 6 16 -Rmod 1 -Rbeg 110.0 -Rend 130.0 -Pbeg 0.0 -Pend 180.0 -Tbeg 0.0 -Tend 180.0 -c contact_color.pdb -p contact.pdb diff.xplor 110.0_130.0.ps
rivem -S 1 -C 6.0 20.0 2.0 3 1 -C -20.0 -6.0 2.0 5 0 -c 5 -v 1 -s 2 -m ncs.def -A 1 -D 0.8 -Rbeg 110.0 -Rend 130.0 -Pbeg 62.0 -Pend 118.0 -Tbeg 55.0 -Tend 95.0 diff.xplor 110_130_average.ps
rivem -A 2 -c 4 -Rmod 3 -dmin -5.0 -dmax 5.0 -l -m ncs.def -v 1 -s 2 -D 0.1 -Pbeg 60.0 -Pend 120 .0 -Tbeg 55.0 -Tend 95.0 -p cva21.pdb cva21_delphi.xplor charge.ps
A. rivem -s 2 -m ncs.def -Rbeg 732.0 -Rend 1020.0 -C 50.0 300.0 25.0 3 1 pbcv_9A.xplor pbcv_r.ps
B. rivem -s 2 -m ncs.def -I -Rbeg 900.0 -Rend 1020.0 -C 90.0 162.0 10.0 3 1 pbcv_9A.xplor pbcv_i.ps
rivem -C -150.0 -0.0 30.0 4 1 -s 2 -sc 12 -ss 10 -sb 0 -sl 7 -m ncs.def -Rbeg 210.0 -Rmod 0 -Pbeg 50.0 -Pend 130.0 -Tbeg 50.0 -Tend 130.0 -c 4 -N 2 60.0 70.0 4 12 16 80.0 115.0 6 10 5 -L 1 60.0 7 0.0 80.0 115.0 0 3 test.xplor -F 1 115.0 70.0 20 0 "Happy Chinese New Year" additional.ps
BUG REPORT: If you meet any bugs or problems of the programs, please email me at cxiao@utep.edu.
 (click to enlarge)
(click to enlarge)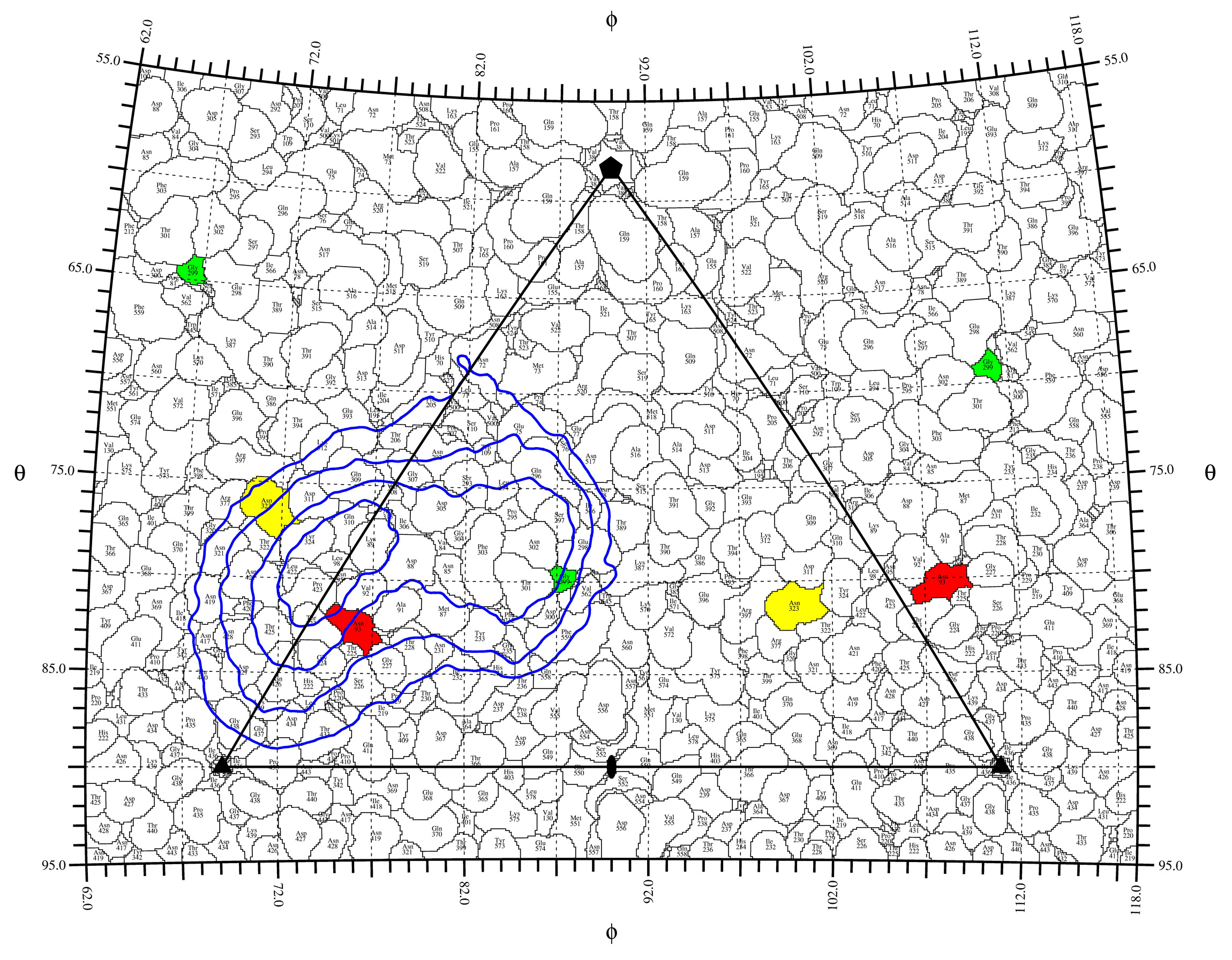 (click to enlarge)
(click to enlarge)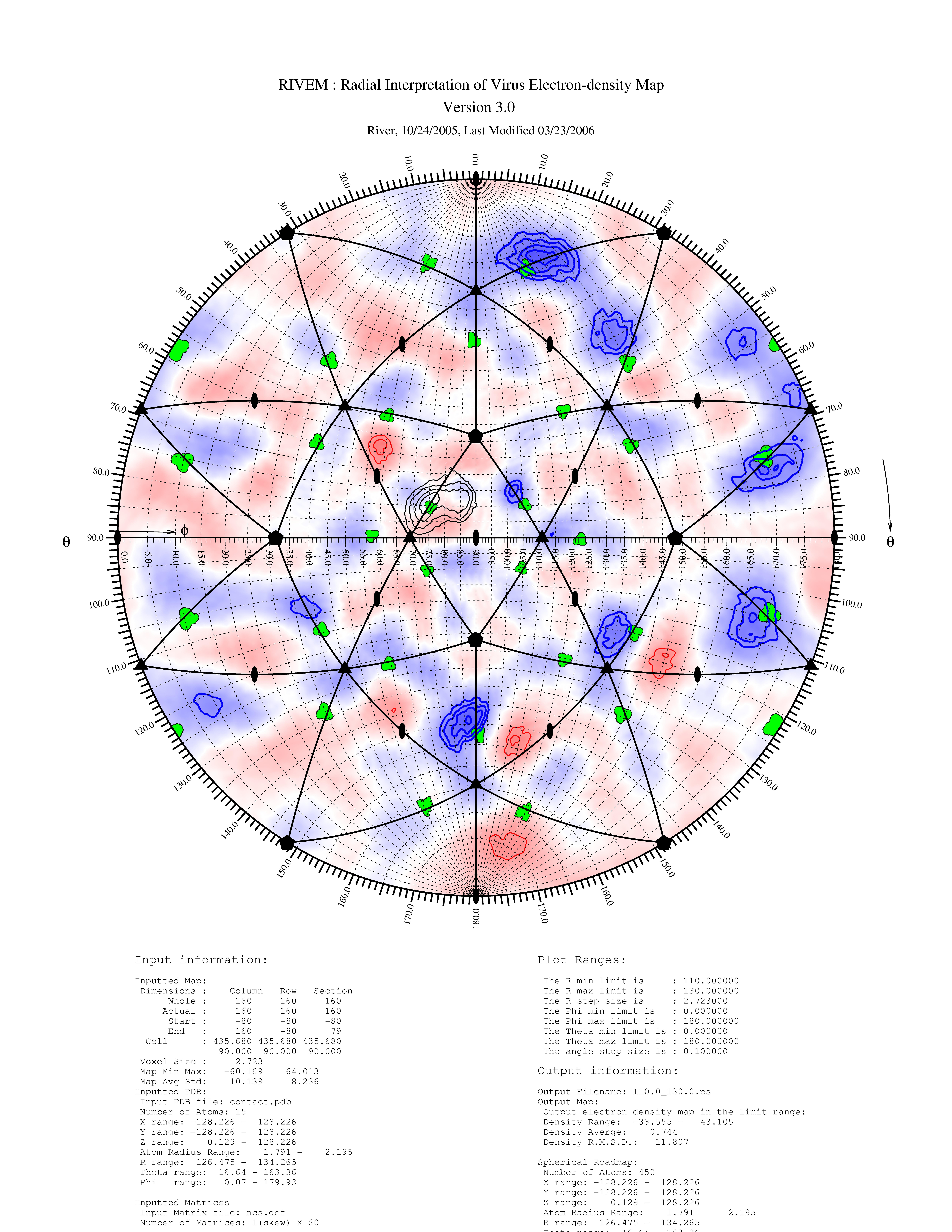 (click to enlarge)
(click to enlarge)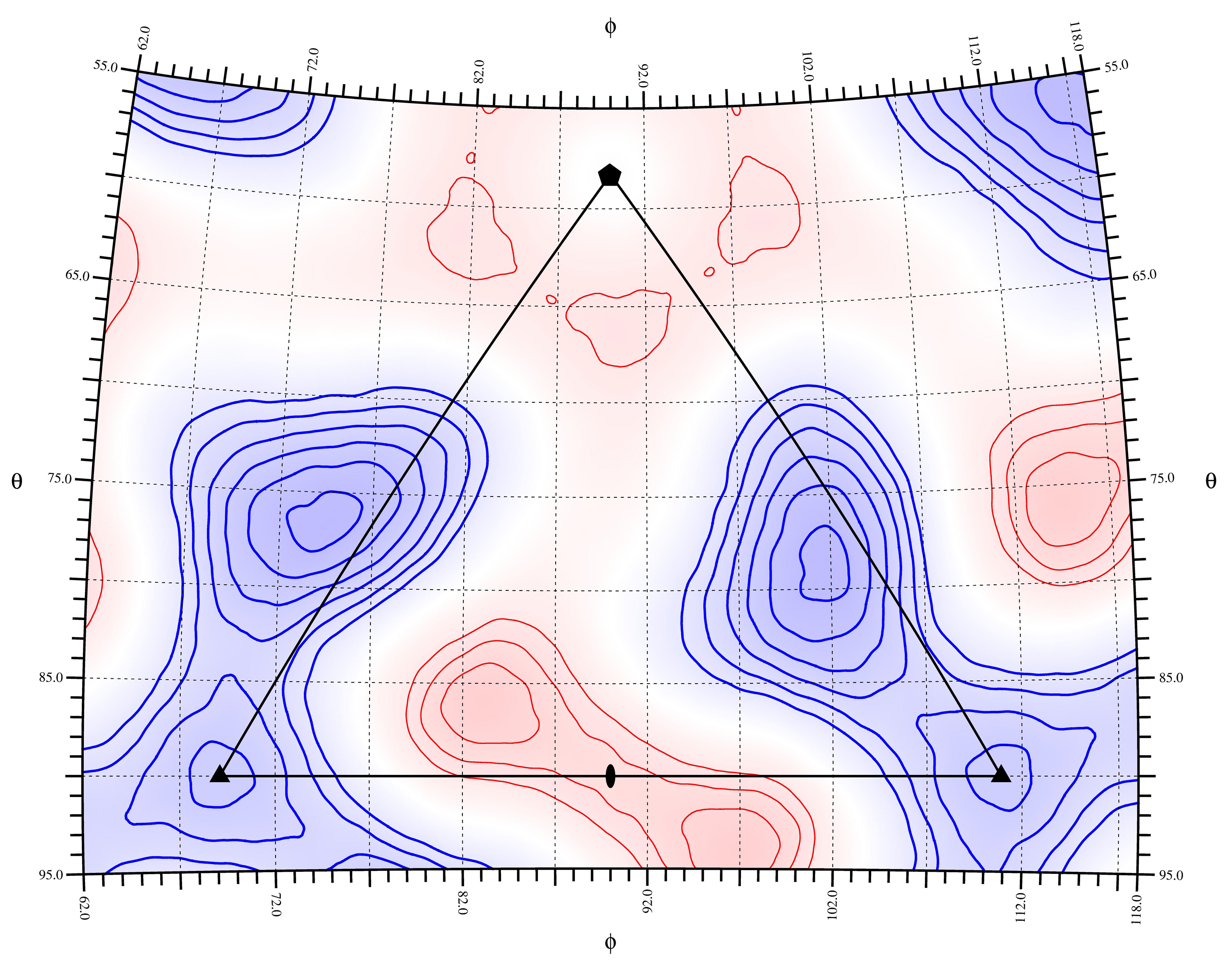 (click to enlarge)
(click to enlarge)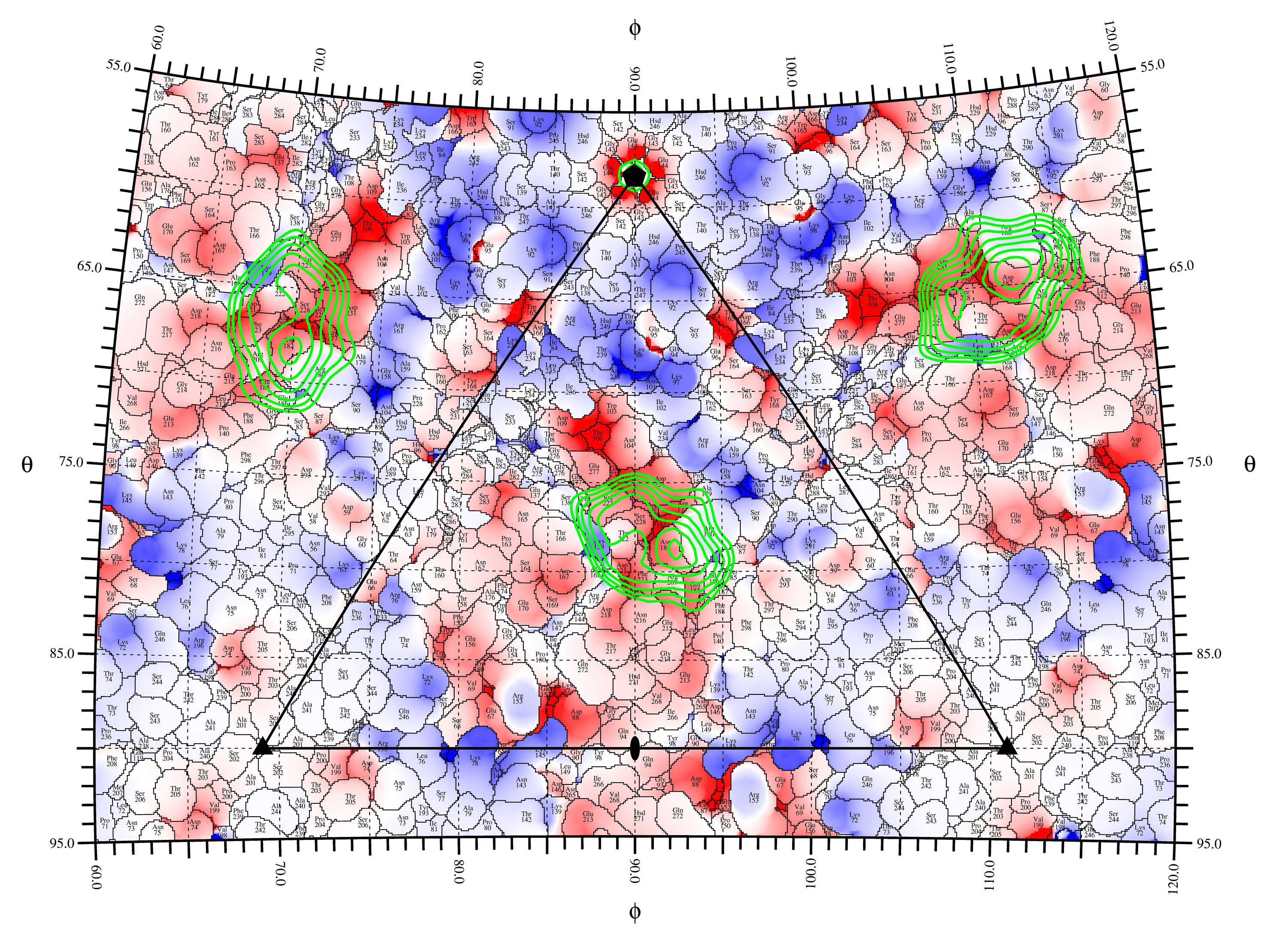 (click to enlarge)
(click to enlarge)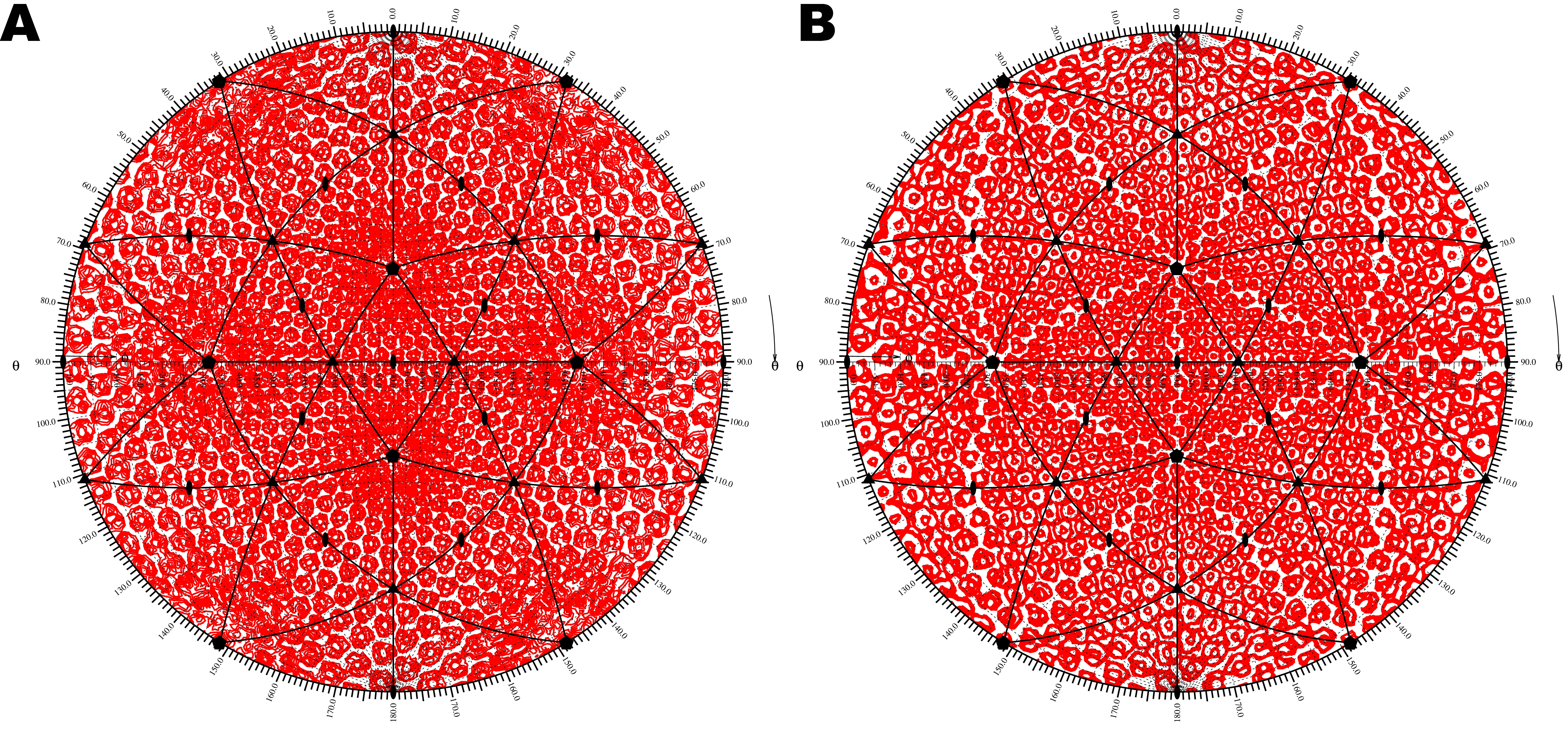 (click to enlarge)
(click to enlarge)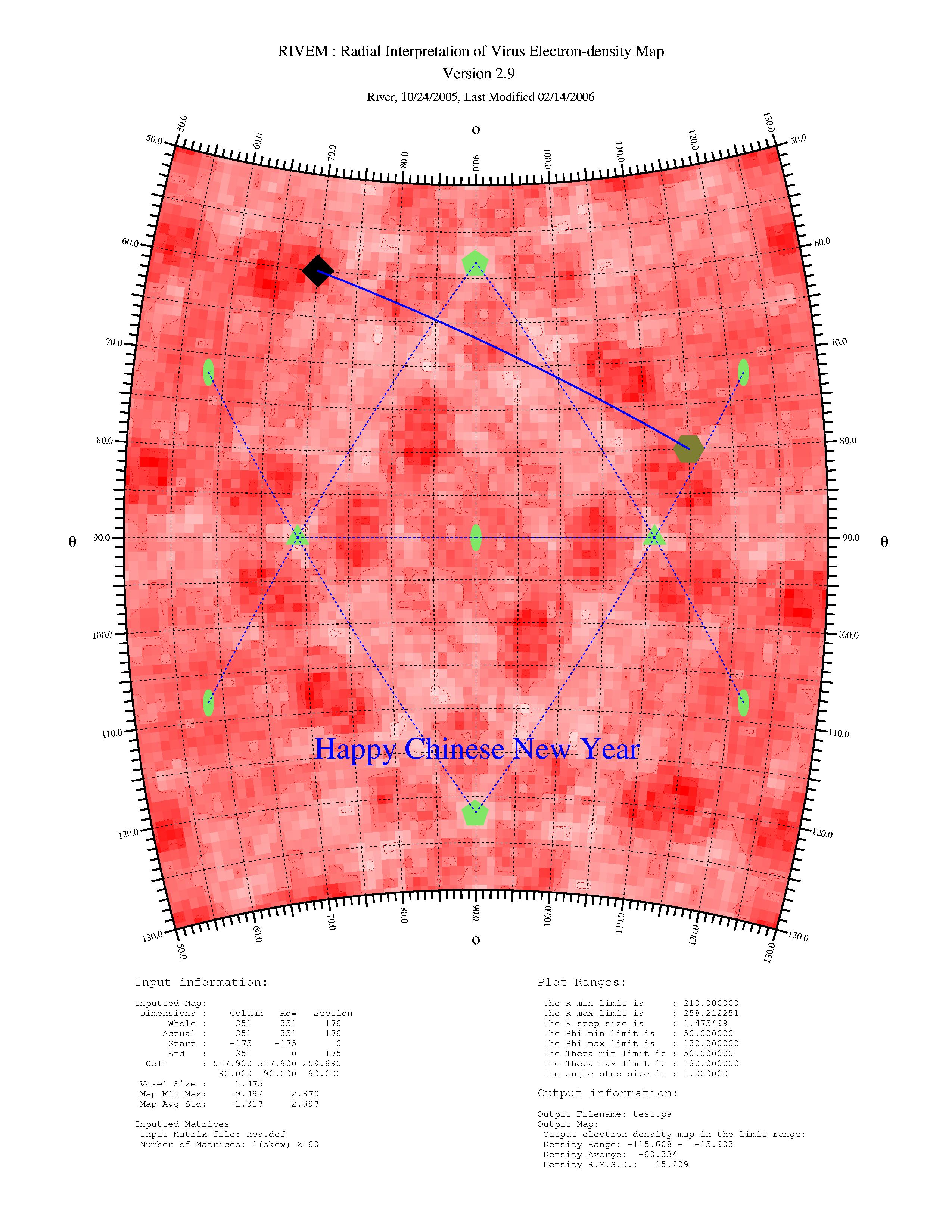 (click to enlarge)
(click to enlarge)